Best Practices for Complex Regulatory Submissions
Development of a novel therapeutic takes several years of intense research and detailed planning. Because target populations are likely to be distributed over multiple regions globally, companies often plan for regulatory submission and approval in multiple countries at the same time, which adds a layer of complexity beyond development of the therapy. The likelihood of successfully launching a new therapy simultaneously in multiple geographic regions depends on thorough planning and disciplined execution from development through approvals.
Organizations need to consider both technical content requirements and company capabilities when developing a global submission strategy. Regulatory agencies and industry experts have collaborated and published harmonized guidelines on the scientific content for regulatory submissions, whether for one or multiple country approvals. Technical teams use this information to conduct appropriate studies and gather critical data to build dossiers that are compliant in several regions at once. However, many organizations are not as adept at factoring in company capabilities while planning for parallel submissions.
Careful planning is critical for success and includes, but is not limited to, factors such as awareness of regulatory, resource, and financial requirements for each submission. IPM’s experience indicates that companies require and value effective planning for specific stages of the submission process, from early stage development through life cycle management.
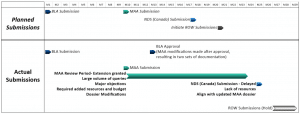
However, regulatory and business operation leads often underestimate the level of planning and technical engagement that agency review periods require. During the period between submission and approval, companies must respond to agency questions and comments as they arise while balancing daily functional responsibilities and other regulatory commitments. A program team may be able to develop a strategy, handle responses, and update the dossier for a single regulatory submission. However, the same team may struggle to execute those activities for several overlapping submissions—especially in multiple countries—with the same rigor. Inadequate planning for overlapping review periods could ultimately lead to resource burnout and missed projections.
RECOMMENDATIONS FOR MANAGING ONGOING, OVERLAPPING SUBMISSIONS
A holistic strategy incorporates the anticipated time, resources, and budget needed to support the review period ahead of submissions and enables teams to effectively manage the workload and mitigate risks. However, many pharmaceutical/biotechnology companies evaluate and establish review period requirements only after submissions are completed.
Over the course of managing such cases, IPM has implemented and recommends the following best practices to maximize product team capabilities.

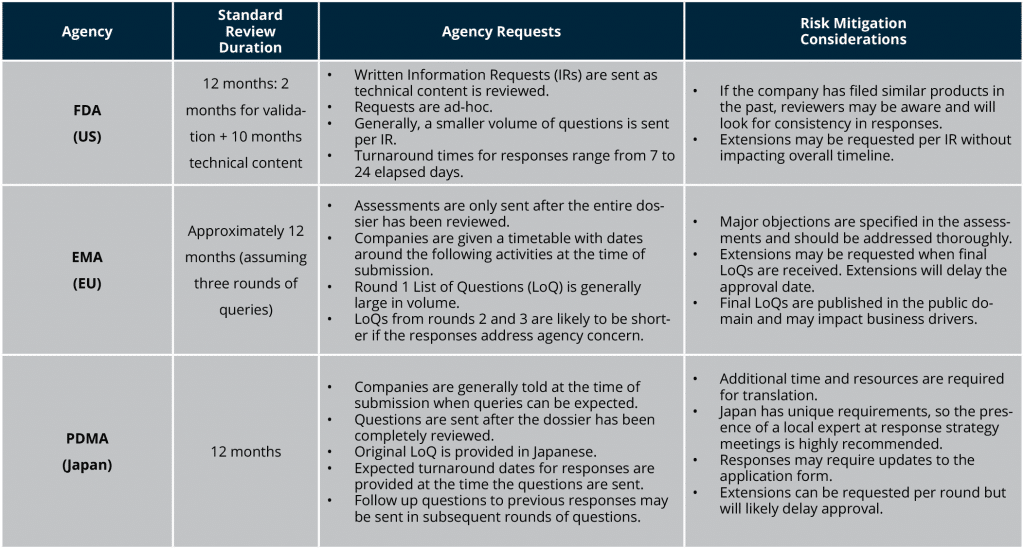
Understand agency requirements and timelines—and how they interact
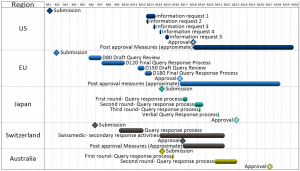
Outline typical agency requests and response times for major market submissions. Ad-hoc agency meetings may be necessary to supply or clarify information. Responses provided during the review cycle can result in updates to the technical content in the original dossier. If the product and regulatory strategy includes overlapping submissions, changes to one filing may impact already approved or subsequent planned submissions, as reviewed material may be made public by agencies. Longer-term solutions proposed as post-approval measures or commitments must be recorded and tracked for timely delivery and may impact one or more submissions.
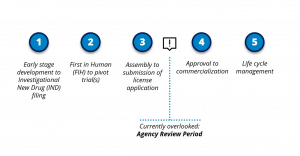
Figure 1: Stages Considered When Developing a Submission Strategy

Assess the dossier and mitigate risk
Objectively review the complete dossier against agency requirements to estimate the volume and scope of expected queries. Identify weak areas or potential gaps that may prompt a major query, multiple post-submission questions, or high-risk post-approval commitments. Consider engaging subject matter experts or advisors to support the risk assessment and suggest strategies to mitigate potential agency concerns.
Classify gaps in the filing based on prior experience with and/or scientific advice from regulatory agencies. Prioritize the high risks and consider staggering submissions so approval in one major market is feasible before pursuing other submissions.
If several medium- to low-risk gaps are identified, consider a strategy that includes submission in one or two major markets simultaneously or with some lag in between; at the same time, pursue extensions or be prepared to withdraw if queries require actions beyond the planned scope or product team capabilities. If query responses are successful, continue with the rest of the global submissions as planned. Document the response strategy and make sure the product team understands it.
If a few low-risk gaps are identified, ensure you have a documented and well-understood response strategy.

Set expectations
Prior to the start of a review period, educate the team on the expected activities and then equip members with the tools and processes that will maximize efficiency. Conduct a kickoff meeting and outline the flow of activities and roles and responsibilities, as well as expectations for management engagement and approvals. Work through potential scenarios requiring varying levels of engagement to pressure test the team’s readiness. For example, major objections from the EMA may result in a decision to conduct additional studies, or the FDA may request quicker response turnaround times than originally planned.

Assign adequate resources
Put a rapid response team in place. Identify which submissions and responses require technical resources to be available continuously and assemble a backup team to manage off-hour requests. Consider allotting resources that are solely responsible for responding to product-specific post-submission requests. Assign roles and responsibilities to each activity, and ensure the team is aware of the expected workload. Consider the teams’ competing priorities and assist them in prioritizing the work.

Implement standard processes and tools
Before the dossier is submitted to an agency, develop and train the submission team(s) on approaches, processes, and use of tools designed to efficiently manage events such as receiving a large volume of queries or ad-hoc information requests, documentation review and revisions, hand-offs, and post-approval commitments.
Incorporate the estimated cost of engaging in review activities when re-evaluating or planning for corporate goals. Companies should assume there will be heavy engagement required to support each submission. If the recalculated expenses are beyond company capabilities, the global submission strategy should be adjusted with a clear evaluation of tradeoffs.
THE BOTTOM LINE
Agency review periods may require a high level of engagement from the company. The learning curve for managing overlapping global review activities can be steep for organizations that overlook this period in the initial planning process. To increase the chances for successful approvals, consider therapeutic needs by region, evaluate submissions to predict queries and mitigate risk, and proactively budget resources and dollars.